Miễn phí
Bệnh giảm gammaglobulin máu Bruton: Chuỗi ca bệnh và tổng quan tài liệu từ Bệnh viện Trung ương King Fahad, Jizan, Ả Rập Xê Út
Bruton’s Agammaglobulinemia: Case Series and Literature Review From King Fahad Central Hospital, Jizan, Saudi Arabia
Ahmed Shamakhi; Nabil Dhayhi; Shatha Matabi; Manal Maashi; Abdullah Dhaifallah Hamdi; Bander Ali; Fadhel Hazazi; Abdullah H. Alhamoud. American Journal of Case Reports. doi: 10.12659/AJCR.949936. Open access.
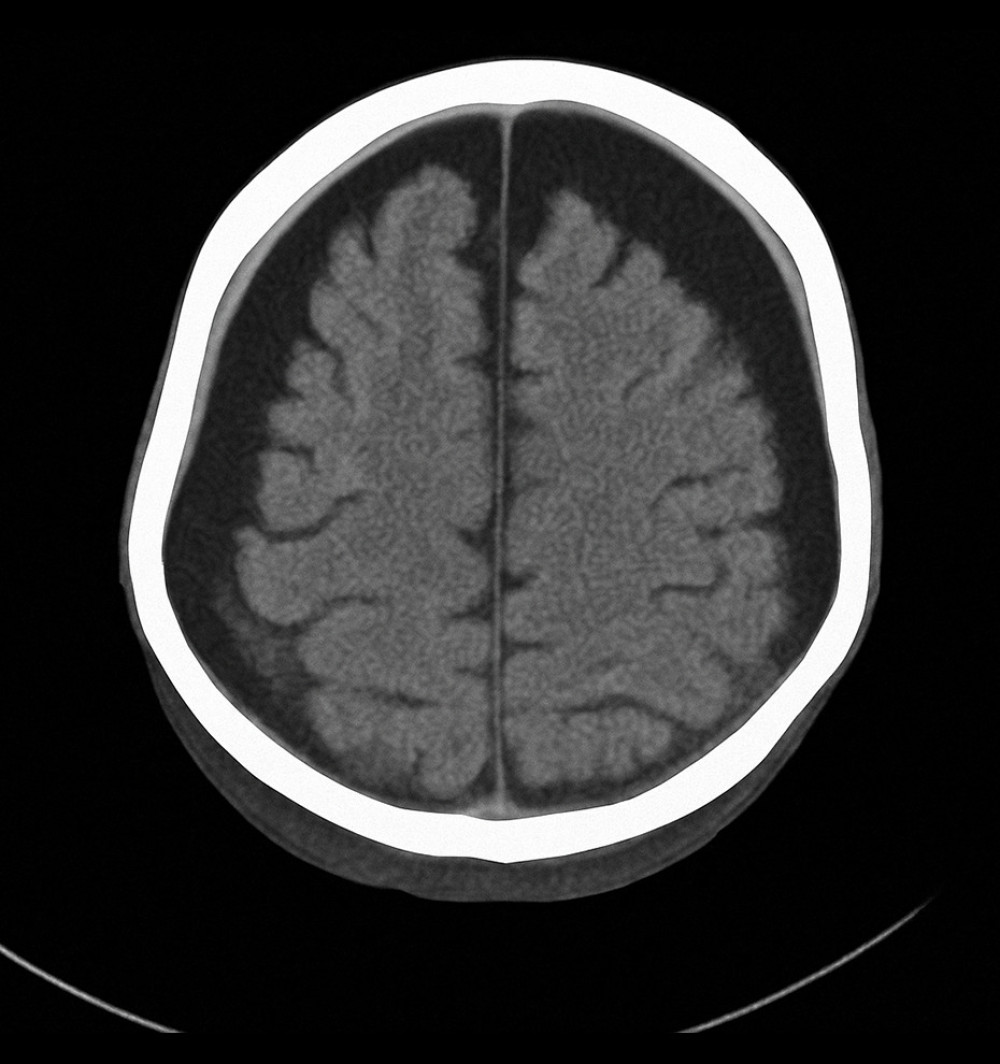
Figure 1
https://jours.isi-science.com/imageXml.php?i=amjcaserep-26-e949936-g001.jpg&idArt=949936&w=1000
Figure 1
Hình 1
Axial computed tomography scan of the brain showing bilateral subdural hygroma and mild cerebral atrophy, consistent with chronic intracranial complications secondary to recurrent infections in X-linked agammaglobulinemia (Case 1).
Hình ảnh chụp cắt lớp vi tính sọ não cắt ngang (axial) cho thấy khối tụ dịch dưới màng cứng hai bên và teo não nhẹ, phù hợp với các biến chứng nội sọ mạn tính thứ phát sau các đợt nhiễm trùng tái phát trong bệnh giảm gammaglobulin máu liên kết nhiễm sắc thể X (Ca bệnh 1).
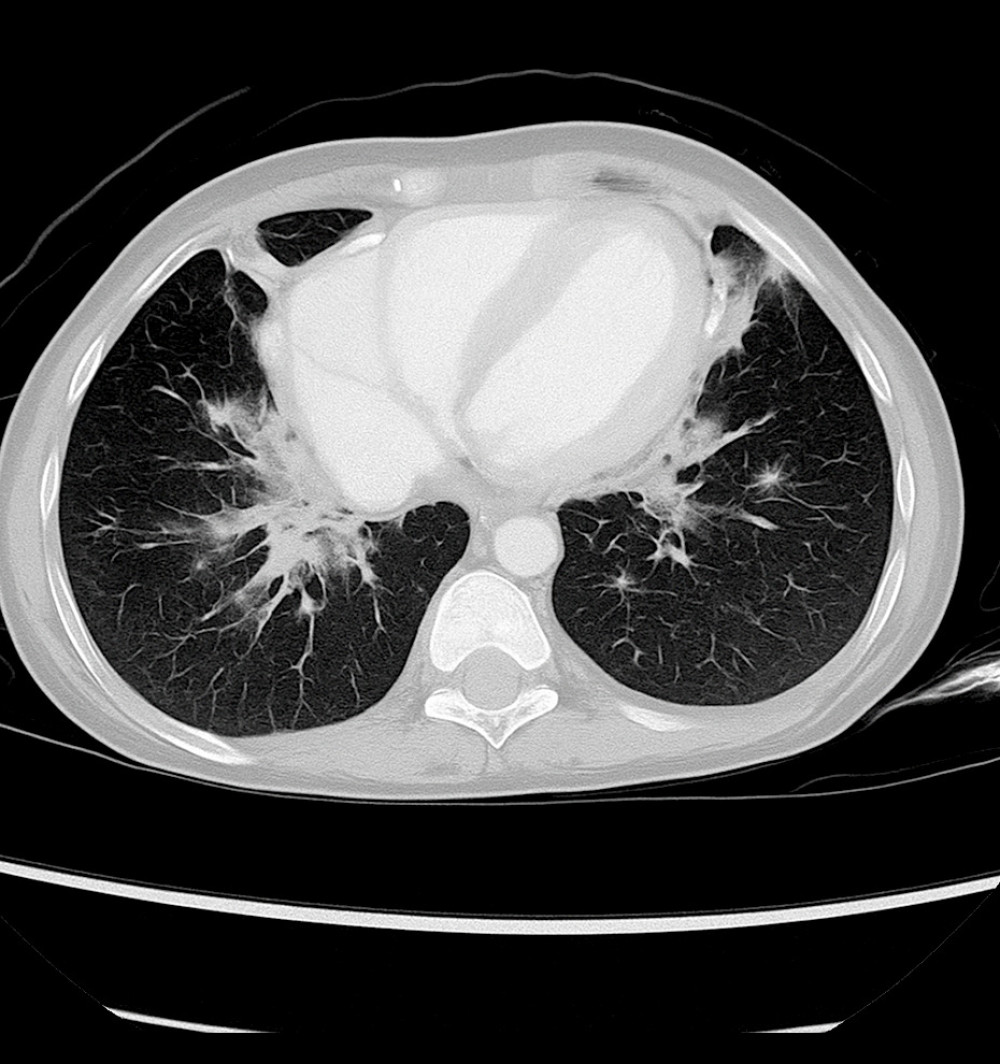
Figure 2
https://jours.isi-science.com/imageXml.php?i=amjcaserep-26-e949936-g002.jpg&idArt=949936&w=1000
Figure 2
Hình 2
Axial chest computed tomography image showing bilateral pulmonary infiltrates with areas of patchy consolidation (Case 2).
Hình ảnh chụp cắt lớp vi tính lồng ngực cắt ngang (axial) cho thấy các tổn thương thâm nhiễm phổi hai bên kèm theo các vùng đông đặc rải rác thành đám (Ca bệnh 2).
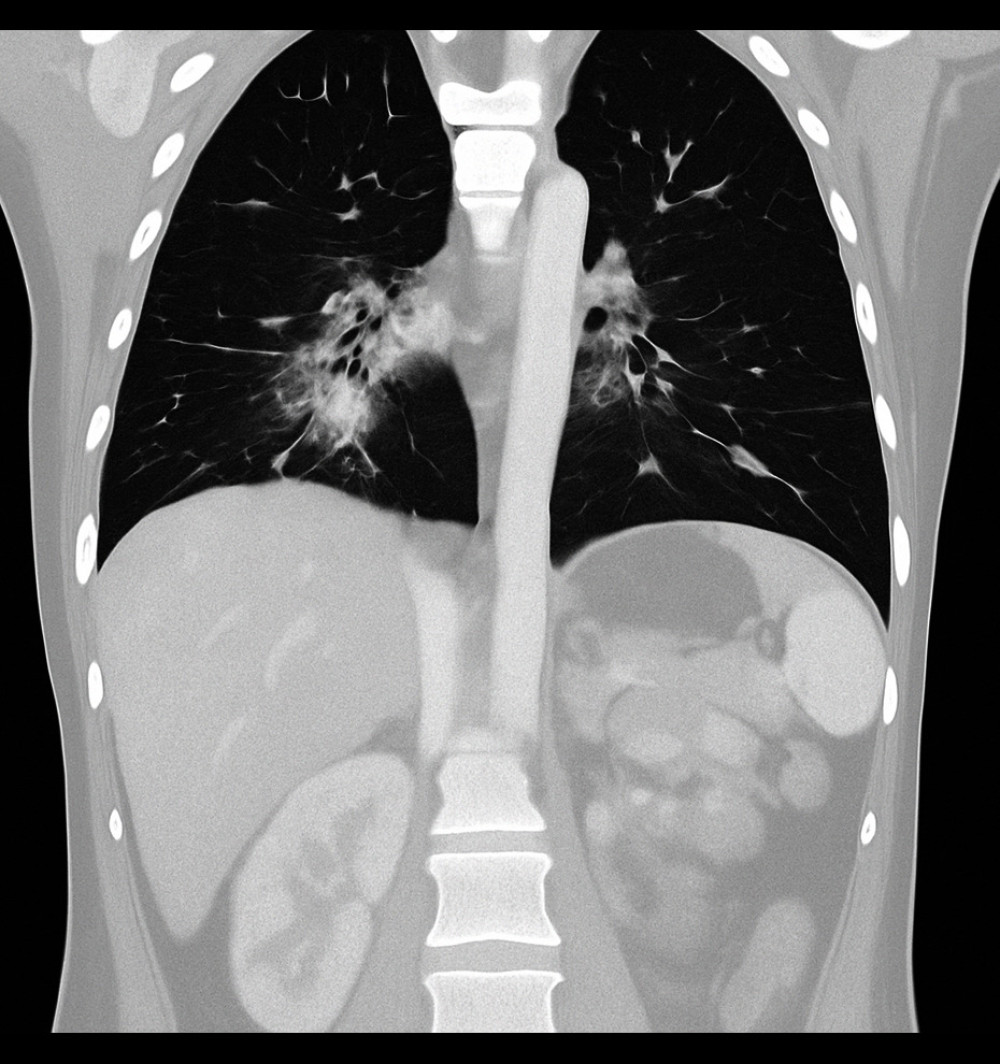
Figure 3
https://jours.isi-science.com/imageXml.php?i=amjcaserep-26-e949936-g003.jpg&idArt=949936&w=1000
Figure 3
Hình 3
Coronal chest computed tomography image showing ground-glass opacities predominantly in the perihilar regions, consistent with an infectious or inflammatory process (Case 2).
Hình ảnh chụp cắt lớp vi tính lồng ngực đứng dọc (coronal) cho thấy các vùng kính mờ chiếm ưu thế ở vùng quanh rốn phổi, phù hợp với diễn tiến nhiễm trùng hoặc viêm (Ca bệnh 2).
Có thể bạn quan tâm
-
Ung thư biểu mô tuyến phổi không triệu chứng kèm di căn tim tại thời điểm chẩn đoán ban đầu: Báo cáo ca bệnh
Asymptomatic Lung Adenocarcinoma With Cardiac Metastasis at Initial Diagnosis: A Case Report
-
Kết quả sống còn của phẫu thuật nội soi lồng ngực hỗ trợ bằng video (VATS) so với phẫu thuật cắt thùy phổi hở cho ung thư phổi: phân tích gộp dữ liệu từng bệnh nhân từ các thử nghiệm ngẫu nhiên
Survival outcome of VATS compared with open lobectomy for lung cancer: an individual patient data meta-analysis of randomised trials
-
Mang thai là cơ hội để thay đổi thói quen ăn uống của gia đình trước khi em bé chào đời
Pregnancy is a chance to reshape family eating habits before the baby arrives