Ca lâm sàng
Đáp ứng với hormone tăng trưởng ở trẻ có đột biến TOMM7 đồng hợp tử: Những góc nhìn điều trị mới
Growth Hormone Response in a Child With a Homozygous TOMM7 Mutation: Novel Therapeutic Insights
Abstract
Tóm tắt
BACKGROUND: Translocase of the outer mitochondrial membrane 7 (TOMM7) encodes a subunit of the mitochondrial translocase complex, which has a critical role in stabilizing the complex and regulating mitochondrial function. Rare individual case reports have identified homozygous TOMM7 mutations associated with Garg-Mishra progeroid syndrome (GMPGS), characterized by dwarfism, facial dysmorphia, developmental delay, and macular scarring. However, few therapeutic interventions have been documented.
BỐI CẢNH: Translocase của màng ngoài ty thể 7 (TOMM7) mã hóa một tiểu đơn vị của phức hợp translocase ty thể, đóng vai trò quan trọng trong việc ổn định phức hợp và điều hòa chức năng ty thể. Một vài báo cáo ca bệnh riêng lẻ hiếm hoi đã xác định các đột biến đồng hợp tử TOMM7 có liên quan đến hội chứng dạng già Garg-Mishra (GMPGS), đặc trưng bởi chứng lùn, biến dạng khuôn mặt, chậm phát triển và sẹo hoàng điểm. Tuy nhiên, có rất ít can thiệp điều trị được ghi nhận.
CASE REPORT: We describe a 2-year-old Han boy from China with severe growth retardation who carries a homozygous TOMM7 mutation (p.Pro29Leu), inherited from consanguineous parents; he has a confirmed diagnosis of GMPGS. Because of growth stagnation, the child has been receiving long-acting recombinant human growth hormone since 31 months of age. After 10 months of treatment, his length increased by 3.8 cm (change in standard deviation score [SDS] of -0.34). This modest decline in SDS sharply contrasted with the precipitous drop of 1.28 SDS during the 10 months before treatment; it represented distinct improvement from the near-complete growth arrest observed between 24 and 31 months of age.
BÁO CÁO CA BỆNH: Chúng tôi mô tả một bé trai 2 tuổi người Hán ở Trung Quốc bị chậm phát triển nghiêm trọng mang đột biến đồng hợp tử TOMM7 (p.Pro29Leu), di truyền từ bố mẹ có quan hệ cận huyết; bệnh nhi đã được chẩn đoán xác định mắc GMPGS. Do tình trạng ngừng tăng trưởng, trẻ đã được điều trị bằng hormone tăng trưởng tái tổ hợp người tác dụng kéo dài từ lúc 31 tháng tuổi. Sau 10 tháng điều trị, chiều dài của trẻ tăng thêm 3,8 cm (thay đổi chỉ số độ lệch chuẩn [SDS] là -0,34). Sự sụt giảm SDS khiêm tốn này tương phản rõ rệt với mức giảm mạnh 1,28 SDS trong 10 tháng trước khi điều trị; điều này thể hiện sự cải thiện rõ rệt so với tình trạng gần như ngừng tăng trưởng hoàn toàn được ghi nhận trong khoảng từ 24 đến 31 tháng tuổi.
CONCLUSIONS: This case highlights the clinical characteristics of children with TOMM7 mutations and offers a potential strategy for managing growth retardation associated with mitochondrial dysfunction.
KẾT LUẬN: Ca bệnh này làm nổi bật các đặc điểm lâm sàng của trẻ mang đột biến TOMM7 và đưa ra một chiến lược tiềm năng trong việc quản lý tình trạng chậm phát triển liên quan đến rối loạn chức năng ty thể.
Keywords: growth disorders, rare diseases, growth hormone
Từ khóa: rối loạn tăng trưởng, bệnh hiếm, hormone tăng trưởng
Introduction
Đặt vấn đề
The TOMM7 gene, also called translocase of the outer mitochondrial membrane 7, encodes a protein with a vital role in mitochondrial function. This protein is part of the translocase of the outer membrane (TOM) complex, which is essential for transporting nuclear-encoded proteins into mitochondria 1. TOMM7 mutations disrupt the mitochondrial protein import process, resulting in mitochondrial dysfunction that manifests as abnormal energy production and protein expression in affected cells 2,3. TOMM7-mutant cells exhibit increased oxygen consumption, suggesting a disconnect between oxidative metabolism and adenosine triphosphate synthesis 4.
Gen TOMM7, còn được gọi là translocase của màng ngoài ty thể 7, mã hóa một loại protein đóng vai trò quan trọng trong chức năng ty thể. Protein này là một phần của phức hợp translocase màng ngoài (TOM), vốn là yếu tố thiết yếu để vận chuyển các protein được mã hóa bởi bộ gen trong nhân vào bên trong ty thể 1. Các đột biến TOMM7 làm gián đoạn quá trình nhập khẩu protein của ty thể, dẫn đến rối loạn chức năng ty thể biểu hiện qua sự sản sinh năng lượng và biểu hiện protein bất thường trong các tế bào bị ảnh hưởng 2,3. Tế bào đột biến TOMM7 cho thấy mức tiêu thụ oxy tăng lên, gợi ý có sự mất kết nối giữa quá trình chuyển hóa oxy hóa và quá trình tổng hợp adenosine triphosphate 4.
A limited number of human diseases have been associated with mutations in genes encoding the subunits of the TOM complex. Homozygous mutations in TOMM7 are related to Garg-Mishra progeroid syndrome (GMPGS), a rare genetic disorder characterized by dwarfism, facial dysmorphia, developmental delay, and macular scarring 3,5. Affected individuals exhibit profound growth impairment. Reported cases have involved progressive postnatal growth retardation, where body height ranges from −4.5 to −8.2 standard deviation scores (SDS) below the mean, underscoring the short stature severity associated with this condition. Furthermore, some patients with TOMM7 mutations also develop moyamoya disease, a cerebrovascular disorder characterized by progressive stenosis of the internal carotid arteries and formation of fragile collateral vessels 4. This condition aligns with findings in animal models, in which TOMM7 deficiency leads to both mitochondrial abnormalities and cerebrovascular defects 6.
Hiện chỉ có một số lượng hạn chế các bệnh ở người được ghi nhận là có liên quan đến các đột biến ở các gen mã hóa cho các tiểu đơn vị của phức hợp TOM. Các đột biến đồng hợp tử ở TOMM7 có liên quan đến hội chứng dạng già Garg-Mishra (GMPGS), một rối loạn di truyền hiếm gặp đặc trưng bởi chứng lùn, biến dạng khuôn mặt, chậm phát triển và sẹo hoàng điểm 3,5. Những cá thể bị ảnh hưởng biểu hiện tình trạng suy giảm tăng trưởng trầm trọng. Các ca bệnh được báo cáo đều liên quan đến tình trạng chậm phát triển tiến triển sau sinh, trong đó chiều cao cơ thể dao động từ −4,5 đến −8,2 điểm lệch chuẩn (SDS) dưới mức trung bình, làm nổi bật mức độ nghiêm trọng của chứng thấp còi liên quan đến tình trạng này. Ngoài ra, một số bệnh nhân có đột biến TOMM7 cũng phát triển bệnh moyamoya, một rối loạn mạch máu não đặc trưng bởi hẹp tiến triển các động mạch cảnh trong và hình thành các mạch máu bàng hệ dễ vỡ 4. Tình trạng này tương thích với các phát hiện trên mô hình động vật, trong đó sự thiếu hụt TOMM7 dẫn đến cả những bất thường về ty thể và các khiếm khuyết mạch máu não 6.
Thus far, few therapeutic interventions have been documented for conditions associated with TOMM7 mutations. Here, we present a Han boy from China with severe growth retardation who carries a homozygous TOMM7 mutation (p.Pro29Leu), inherited from consanguineous parents. Growth hormone therapy promoted further increases in height, indicating that this treatment could potentially alleviate the growth retardation caused by TOMM7 deficiency. To our knowledge, this report describes the first application of growth hormone therapy in a patient with a TOMM7 mutation, providing an opportunity to evaluate its potential as a treatment strategy.
Cho đến nay, có rất ít can thiệp điều trị được ghi nhận đối với các tình trạng liên quan đến đột biến TOMM7. Tại đây, chúng tôi trình bày một bé trai người Hán ở Trung Quốc bị chậm phát triển nghiêm trọng mang đột biến đồng hợp tử TOMM7 (p.Pro29Leu), di truyền từ bố mẹ có quan hệ cận huyết. Liệu pháp hormone tăng trưởng đã thúc đẩy chiều cao tăng thêm, cho thấy phương pháp điều trị này có tiềm năng làm giảm nhẹ tình trạng chậm phát triển do thiếu hụt TOMM7 gây ra. Theo hiểu biết của chúng tôi, báo cáo này mô tả ứng dụng đầu tiên của liệu pháp hormone tăng trưởng ở một bệnh nhân có đột biến TOMM7, mang lại cơ hội để đánh giá tiềm năng của nó như một chiến lược điều trị.
Case Report
Báo cáo ca bệnh
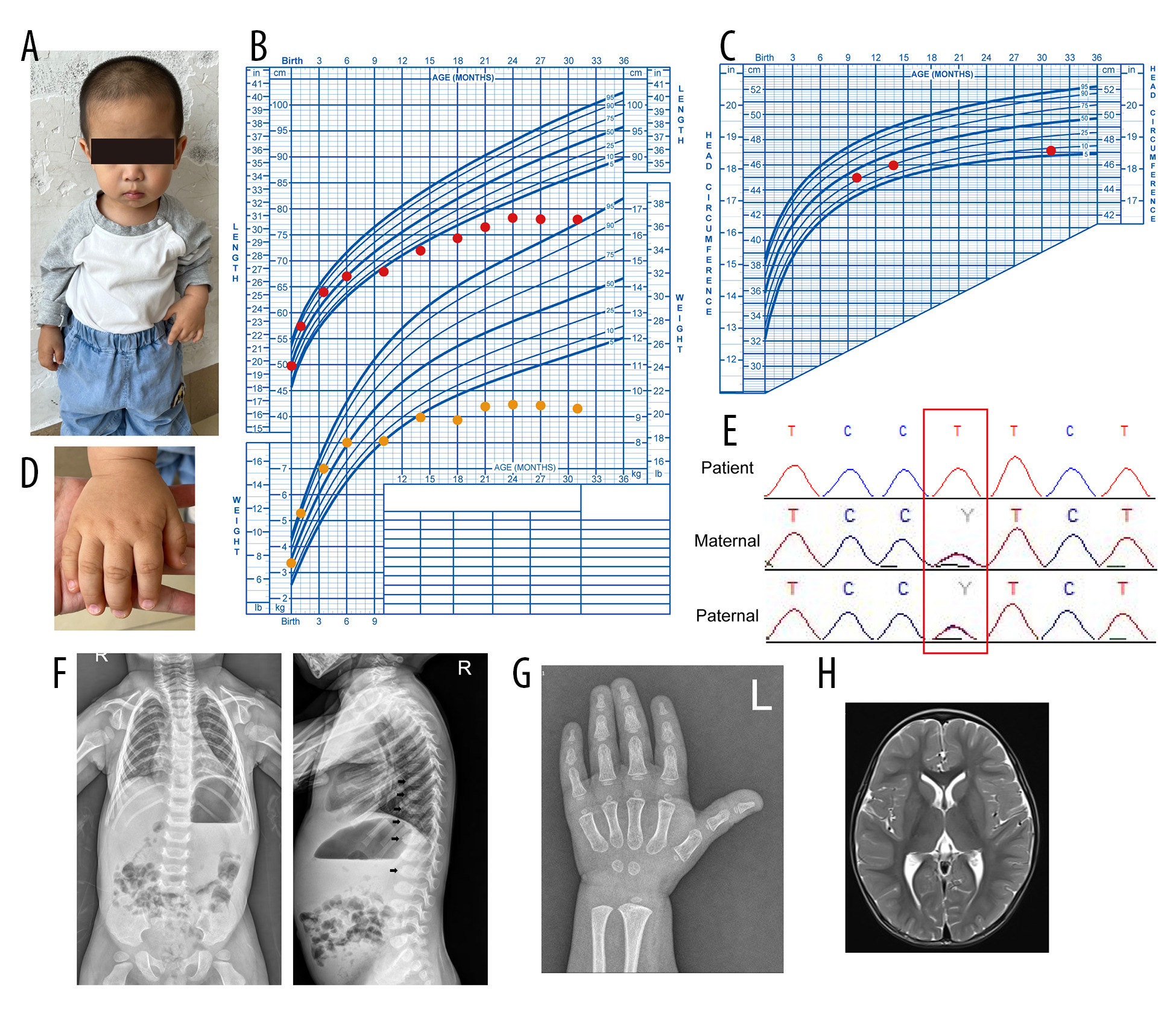
A Han Chinese boy first visited the clinic at 31 months of age for treatment of severe growth retardation that had persisted for at least 25 months (Figure 1A). He was born through a normal full-term delivery to consanguineous parents and had no congenital deformities. He was the second child, with 1 sibling – a 7-year-old brother who showed no clinical manifestations of growth retardation. At birth, he weighed 3,200 g (25th–50th percentile) and measured 50 cm (50th–75th percentile); he had no evident congenital anomalies. There was no prior medical history and no record of medications. His mother had exhibited good health throughout the pregnancy.
Một bé trai người Hán lần đầu tiên đến khám tại phòng khám lúc 31 tháng tuổi để điều trị tình trạng chậm phát triển nghiêm trọng đã kéo dài ít nhất 25 tháng (Figure 1A). Trẻ sinh đủ tháng, sinh thường, là con của bố mẹ có quan hệ cận huyết và không có dị tật bẩm sinh. Trẻ là con thứ hai trong gia đình, có 1 anh trai 7 tuổi không biểu hiện triệu chứng lâm sàng của tình trạng chậm phát triển. Lúc sinh, trẻ nặng 3.200 g (bách phân vị thứ 25–50) và dài 50 cm (bách phân vị thứ 50–75); không phát hiện bất thường bẩm sinh rõ rệt nào. Trẻ không có tiền sử bệnh lý trước đó và không có bệnh án sử dụng thuốc. Người mẹ có sức khỏe tốt trong suốt thai kỳ.
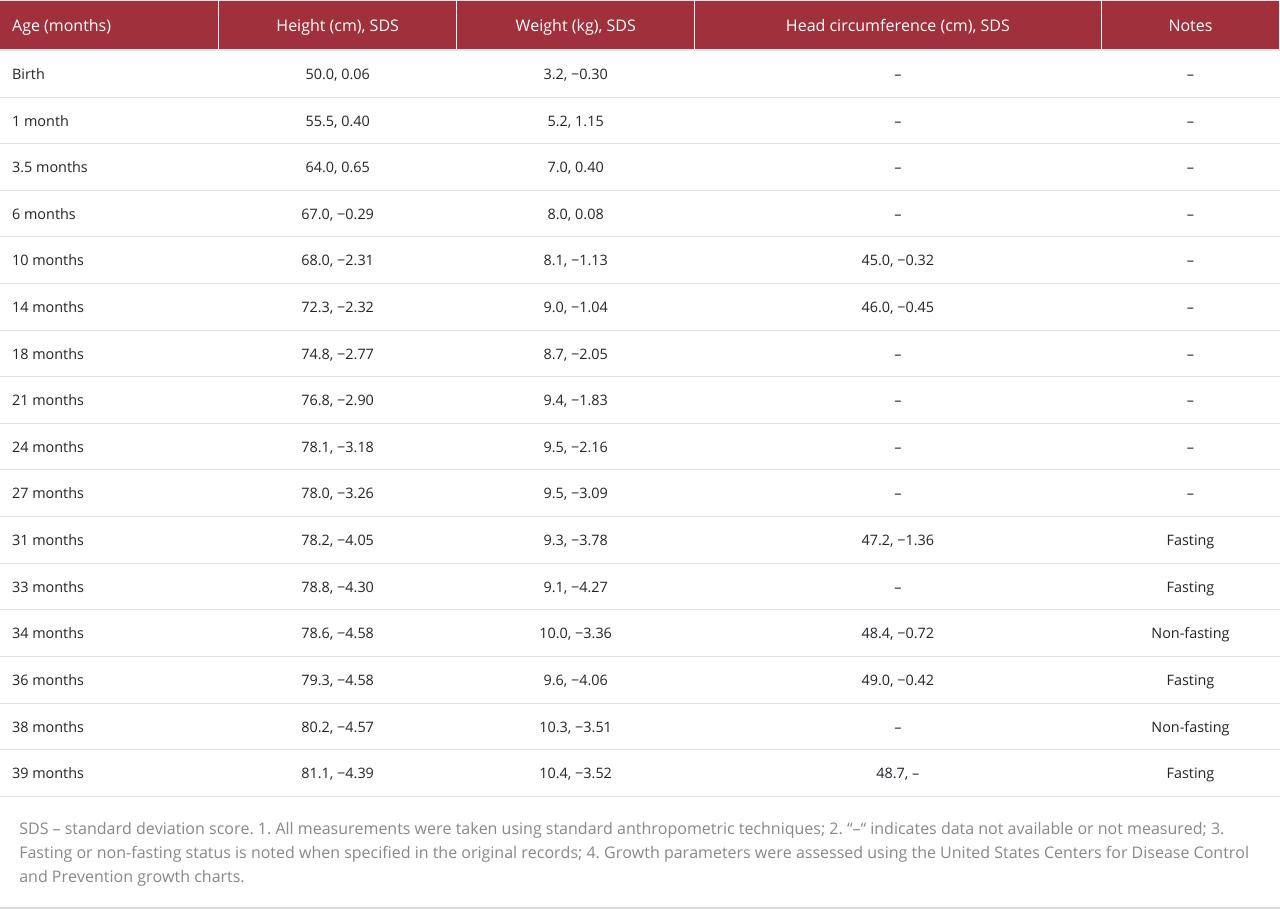
The child experienced substantial delays in length and weight beginning at 6 months of age (Figure 1B, Table 1), which prompted close monitoring and intervention at the primary hospital. During this period, his parents received comprehensive guidance regarding his nutrition and diet, including specific recommendations to increase energy intake for healthy growth. Despite these efforts and individualized dietary strategies, there was no meaningful improvement in his growth trajectory. By 24 months, he exhibited stagnation in length and weight gain (Figure 1B), but his head circumference remained normal (Figure 1C). These observations led to concerns about the underlying factors contributing to his lack of progress and triggered a thorough evaluation of his overall health and nutrition.
Trẻ bắt đầu bị chậm phát triển đáng kể cả về chiều dài và cân nặng từ lúc 6 tháng tuổi (Figure 1B, Table 1), điều này đã thúc đẩy việc theo dõi chặt chẽ và can thiệp tại bệnh viện tuyến cơ sở. Trong giai đoạn này, bố mẹ trẻ đã nhận được hướng dẫn toàn diện về dinh dưỡng và chế độ ăn uống, bao gồm các khuyến nghị cụ thể nhằm tăng lượng năng lượng dung nạp để tăng trưởng khỏe mạnh. Bất chấp những nỗ lực này và các chiến lược ăn uống được cá nhân hóa, biểu đồ tăng trưởng của trẻ vẫn không có sự cải thiện ý nghĩa nào. Đến lúc 24 tháng tuổi, trẻ biểu hiện tình trạng ngừng tăng trưởng về cả chiều dài và cân nặng (Figure 1B), nhưng chu vi vòng đầu vẫn bình thường (Figure 1C). Những quan sát này dẫn đến những lo ngại về các yếu tố tiềm ẩn góp phần vào việc chậm tiến triển của trẻ và kích hoạt một cuộc đánh giá toàn diện về sức khỏe tổng quát và dinh dưỡng của trẻ.
The patient displayed several additional clinical features, including broad hands with a distinctive shape and small, dystrophic nails (Figure 1D). He also exhibited nystagmus and muscle hypotonia, which affected mobility and coordination. Ophthalmologic examination revealed bilateral congenital macular defects and amblyopia in both eyes. His mental development showed slight delays in motor and language skills. Neurodevelopmental assessment at 27 months of age, conducted using the Griffiths Developmental Scale-Chinese version, revealed mild global developmental delay in gross motor, personal-social, auditory-linguistic, and visual-expressive domains (at the 7.5th percentile). However, there was no evidence of the distinctive facial features identified in previous cases, including low-set and protruding ears, a beaked nose, and micrognathia. The patient’s head circumference remained at approximately the 25th percentile for age.
Bệnh nhi có một số đặc điểm lâm sàng khác, bao gồm bàn tay rộng với hình dạng đặc trưng và các móng tay nhỏ, loạn dưỡng (Figure 1D). Trẻ cũng biểu hiện chứng rung giật nhãn cầu và giảm trương lực cơ, gây ảnh hưởng đến khả năng vận động và phối hợp động tác. Khám mắt phát hiện khuyết tật hoàng điểm bẩm sinh hai bên và nhược thị ở cả hai mắt. Phát triển tâm thần của trẻ cho thấy sự chậm trễ nhẹ về các kỹ năng vận động và ngôn ngữ. Đánh giá phát triển thần kinh lúc 27 tháng tuổi, được thực hiện bằng Thang đo Phát triển Griffiths – phiên bản tiếng Trung, cho thấy trẻ bị chậm phát triển toàn diện mức độ nhẹ trong các lĩnh vực vận động thô, cá nhân – xã hội, thính giác – ngôn ngữ, và thị giác – biểu cảm (ở bách phân vị thứ 7.5). Tuy nhiên, không có bằng chứng nào về các đặc điểm khuôn mặt đặc trưng từng được xác định trong các ca bệnh trước đây, bao gồm tai đóng thấp và vểnh, mũi khoằm và cằm nhỏ. Chu vi vòng đầu của bệnh nhi vẫn duy trì ở mức khoảng bách phân vị thứ 25 so với tuổi.
Whole-exome sequencing revealed that the child carried a homozygous TOMM7 gene variant (NM_019059.5: Exon 1: c.86C>T [p.Pro29Leu]), inherited from both parents. Post hoc re-evaluation using American College of Medical Genetics and Genomics guidelines classified this variant as “likely pathogenic” (PS3 + PM2_Supporting + PM3_Supporting). DNA from the patient and his parents was isolated and subjected to Sanger sequencing to confirm the mutation (Figure 1E). The results demonstrated that both parents were heterozygous for the mutation. Further sequencing revealed that the patient’s sibling did not inherit this genetic variant.
Kết quả giải trình tự vùng mã hóa toàn bộ (whole-exome sequencing) cho thấy trẻ mang một biến thể gen TOMM7 đồng hợp tử (NM_019059.5: Exon 1: c.86C>T [p.Pro29Leu]), di truyền từ cả bố và mẹ. Đánh giá lại sau đó theo hướng dẫn của Trường Di truyền học và Bộ gen Y học Hoa Kỳ (ACMG) đã phân loại biến thể này là “có thể gây bệnh” (PS3 + PM2_Supporting + PM3_Supporting). DNA của bệnh nhi và bố mẹ đã được tách chiết và thực hiện giải trình tự Sanger để xác nhận đột biến (Figure 1E). Các kết quả chứng minh rằng cả bố và mẹ đều dị hợp tử đối với đột biến này. Kết quả giải trình tự thêm cho thấy anh trai của bệnh nhi không di truyền biến thể di truyền này.
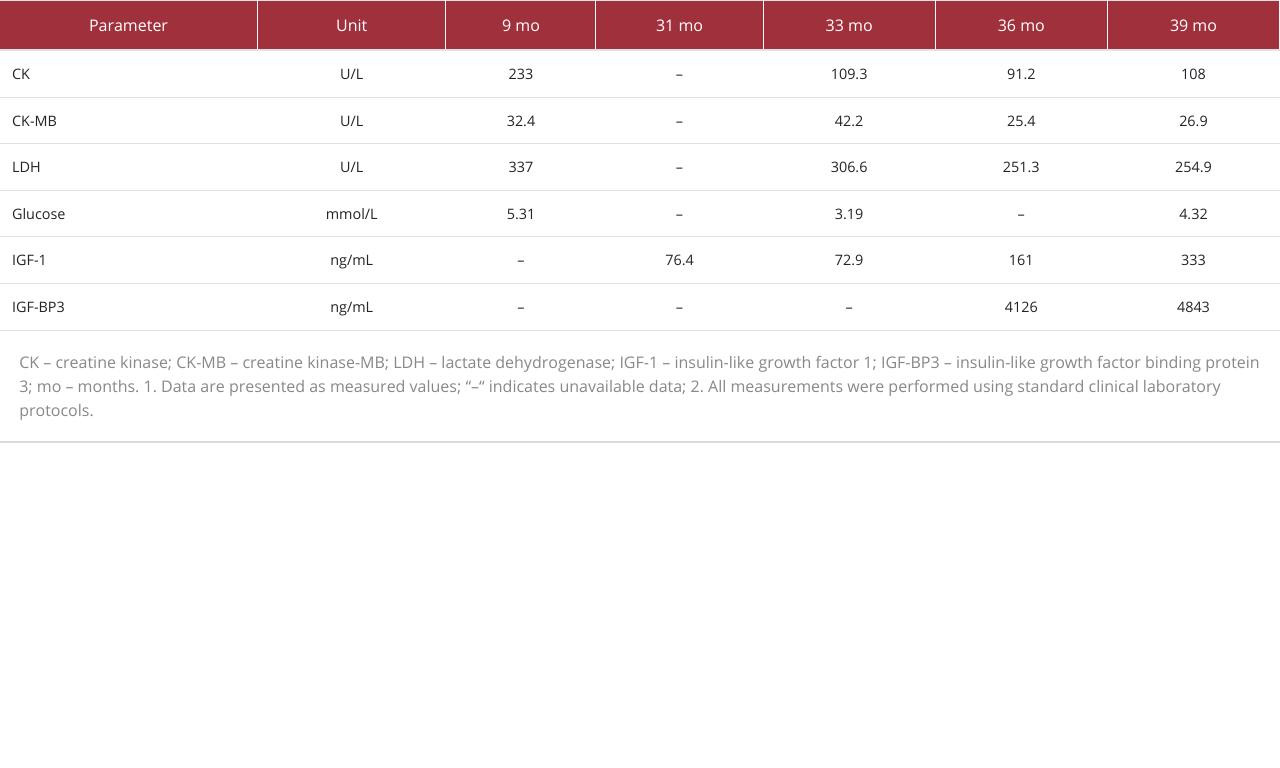
Laboratory tests, including complete blood count; hepatic and renal function; and serum levels of sodium, potassium, chloride, calcium, phosphorus, magnesium, urea, creatinine, total protein, albumin, globulin, bilirubin, lipids, 25-hydroxyvitamin D, tumor markers, and allergens, all showed results within normal limits. However, slightly elevated serum levels of lactate dehydrogenase (LDH) and creatine kinase-MB (CK-MB) were observed (Table 2). Endocrine evaluation revealed normal thyroid function, adrenocorticotropic hormone, cortisol, and sex hormone levels. The serum level of insulin-like growth factor 1 (IGF-1) was at the lower limit of the normal range. A growth hormone stimulation test indicated partial growth hormone deficiency.
Các xét nghiệm phòng thí nghiệm, bao gồm tổng phân tích tế bào máu ngoại vi; chức năng gan và thận; và nồng độ trong huyết thanh của natri, kali, clorua, canxi, phốt pho, magiê, urê, creatinine, protein toàn phần, albumin, globulin, bilirubin, lipid, 25-hydroxyvitamin D, các dấu ấn khối u và các dị nguyên, đều cho kết quả trong giới hạn bình thường. Tuy nhiên, nồng độ lactate dehydrogenase (LDH) và creatine kinase-MB (CK-MB) trong huyết thanh tăng nhẹ đã được ghi nhận (Table 2). Đánh giá nội tiết cho thấy chức năng tuyến giáp, hormone vỏ thượng thận (ACTH), cortisol và nồng độ hormone sinh dục bình thường. Nồng độ yếu tố tăng trưởng tương tự insulin 1 (IGF-1) trong huyết thanh ở mức giới hạn dưới của khoảng bình thường. Nghiệm pháp kích thích hormone tăng trưởng cho thấy tình trạng thiếu hụt hormone tăng trưởng một phần.
X-ray imaging revealed anterior vertebral beaking, resembling mucopolysaccharide-related bony changes in the spine (Figure 1F). Cardiac ultrasound showed mild regurgitation of the bicuspid and tricuspid valves. Ultrasound examinations of the liver, biliary system, pancreas, spleen, kidneys, adrenal glands, and retroperitoneum revealed no clinically significant abnormalities. Bone age, assessed at 2 years and 2 months, showed results consistent with chronological age (Figure 1G). Brain magnetic resonance imaging revealed no signs of ischemia or hemorrhage. Magnetic resonance arteriography was not performed (Figure 1H).
Hình ảnh X-quang cho thấy có hình mỏ chim ở phía trước thân đốt sống, giống với những thay đổi xương liên quan đến bệnh mucopolysaccharide ở cột sống (Figure 1F). Siêu âm tim cho thấy hở nhẹ van hai lá và van ba lá. Siêu âm gan, hệ mật, tụy, lách, thận, tuyến thượng thận và khoang sau phúc mạc không phát hiện bất thường nào có ý nghĩa lâm sàng. Tuổi xương, được đánh giá lúc 2 tuổi 2 tháng, cho kết quả phù hợp với tuổi thực (Figure 1G). Chụp cộng hưởng từ não không phát hiện dấu hiệu thiếu máu cục bộ hay xuất huyết. Chụp cộng hưởng từ mạch máu não đã không được thực hiện (Figure 1H).
Because the growth hormone stimulation test indicated partial growth hormone deficiency, long-acting recombinant human growth hormone (hGH, Jintrolong®) therapy was initiated at 31 months of age. The initial dosage was 1.5 mg once weekly (0.16 mg·kg−1·wk−1). After 3 months of therapy, the dose was increased to 1.8 mg/week (0.18 mg·kg−1·wk−1); it was titrated up to a maintenance dose of 2.0 mg/week (0.20 mg·kg−1·wk−1) after 7 months, which has since been maintained. After 10 months of treatment, the child’s growth showed improvement. His length increased to 82.0 cm (an increase of 3.8 cm, with a change in standard deviation score [ΔSDS] of −0.34), and his weight reached 10.4 kg (an increase of 1.1 kg, ΔSDS of +0.26). This modest decline in length SDS sharply contrasted with the precipitous drop of 1.28 SDS (from −2.77 to −4.05) observed during the 10-month period before treatment and represented substantial improvement from the near-complete growth arrest documented between 24 and 31 months of age (Table 1). The positive change in weight SDS further underscored the overall benefit of the therapy. Serum IGF-1 increased from 76.4 ng/mL to 161 ng/mL, and IGF-binding protein-3 (BP3) reached 4126 ng/mL. Blood tests for glucose, glycosylated hemoglobin, insulin, liver and renal function, lipids, and electrolytes revealed no abnormalities (Table 2).
Vì nghiệm pháp kích thích hormone tăng trưởng chỉ ra tình trạng thiếu hụt hormone tăng trưởng một phần, liệu pháp hormone tăng trưởng tái tổ hợp người tác dụng kéo dài (hGH, Jintrolong®) đã được bắt đầu lúc 31 tháng tuổi. Liều ban đầu là 1,5 mg mỗi tuần một lần (0,16 mg·kg−1·wk−1). Sau 3 tháng điều trị, liều được tăng lên 1,8 mg/tuần (0,18 mg·kg−1·wk−1); liều lượng được chuẩn độ tăng dần đến liều duy trì 2,0 mg/tuần (0,20 mg·kg−1·wk−1) sau 7 tháng và được duy trì từ đó đến nay. Sau 10 tháng điều trị, sự tăng trưởng của trẻ đã có cải thiện. Chiều dài của trẻ tăng lên 82,0 cm (tăng thêm 3,8 cm, với mức thay đổi chỉ số độ lệch chuẩn [ΔSDS] là −0.34), và cân nặng của trẻ đạt 10,4 kg (tăng thêm 1,1 kg, ΔSDS là +0.26). Sự sụt giảm khiêm tốn về chỉ số SDS chiều dài này tương phản rõ rệt với mức giảm mạnh 1,28 SDS (từ −2.77 xuống −4.05) được ghi nhận trong khoảng thời gian 10 tháng trước khi điều trị, và thể hiện sự cải thiện đáng kể so với tình trạng gần như ngừng tăng trưởng hoàn toàn được ghi nhận giữa tháng thứ 24 và 31 (Table 1). Sự thay đổi tích cực về chỉ số SDS cân nặng càng khẳng định thêm lợi ích đồng hóa tổng thể của liệu pháp này. Nồng độ IGF-1 trong huyết thanh tăng từ 76,4 ng/mL lên 161 ng/mL, và protein-3 gắn hormone tăng trưởng tương tự insulin (IGF-BP3) đạt 4126 ng/mL. Các xét nghiệm máu về glucose, hemoglobin glycosylated (HbA1c), insulin, chức năng gan và thận, lipid và điện giải đồ không phát hiện bất thường nào (Table 2).
Discussion
Bàn luận
We have described a boy with a homozygous missense mutation in TOMM7 (p.Pro29Leu) inherited from consanguineous parents. He exhibited normal growth parameters at birth but developed substantial growth delay beginning at 6 months of age, followed by stagnation at 2 years of age, mild developmental delays, fundoscopic macular defects, and specific skeletal changes. We found that growth hormone therapy may serve as a potential treatment to improve growth retardation. To our knowledge, this is the first report concerning the use of growth hormone therapy in a patient with a homozygous TOMM7 mutation.
Chúng tôi đã mô tả một bé trai mang đột biến đột biến sai nghĩa đồng hợp tử trong gen TOMM7 (p.Pro29Leu) di truyền từ bố mẹ có quan hệ cận huyết. Bệnh nhi biểu hiện các chỉ số tăng trưởng bình thường khi sinh nhưng bắt đầu bị chậm phát triển đáng kể từ lúc 6 tháng tuổi, tiếp theo là tình trạng ngừng tăng trưởng lúc 2 tuổi, chậm phát triển nhẹ, khuyết tật hoàng điểm khi soi đáy mắt và các thay đổi đặc hiệu ở xương. Chúng tôi nhận thấy rằng liệu pháp hormone tăng trưởng có thể đóng vai trò như một phương pháp điều trị tiềm năng để cải thiện tình trạng chậm phát triển thể chất. Theo hiểu biết của chúng tôi, đây là báo cáo đầu tiên liên quan đến việc sử dụng liệu pháp hormone tăng trưởng ở một bệnh nhân có đột biến TOMM7 đồng hợp tử.
TOMM7 encodes a subunit of the translocase of the outer mitochondrial membrane. The TOMM7 protein regulates the assembly and stability of the translocase complex. Few cases of TOMM7 gene mutations have been reported because symptoms require a homozygous mutation; heterozygous individuals only carry the variant and exhibit no abnormal clinical features. To date, only 12 cases of TOMM7 mutations have been documented, including 2 pathogenic missense variants (c.73T>C, p.Trp25Arg 5; c.86C>T, p.Pro29Leu 3) and a biallelic splicing variant (c.153-2A>C 7). Short stature and developmental delay are the most common symptoms associated with TOMM7 deficiency caused by hypomorphic variants such as c.73T>C, p.Trp25Arg 5. Biallelic loss-of-function variants that disrupt TOMM complex activity cause severe mitochondrial dysfunction and result in early-onset Leigh syndrome 7. Among the 12 reported cases, 1 involved a man of Chinese descent living in Malaysia, 9 were Taiwanese, and the remaining 2 were from Japan and India. Our report involves a boy from a Han Chinese family with the TOMM7 p.Pro29Leu variant; all known patients have been of Asian ancestry. This TOMM7 variant also shows considerably higher allele frequency in East Asian populations (0.000652, 12/18 394 alleles) compared with the global aggregate (0.000048, 12/251 470 alleles) in the Genome Aggregation Database (gnomAD). Potential ethnic variance in GMPGS incidence requires further investigation.
TOMM7 mã hóa một tiểu đơn vị của phức hợp translocase của màng ngoài ty thể. Protein TOMM7 điều hòa sự lắp ráp và tính ổn định của phức hợp translocase. Rất ít ca bệnh đột biến gen TOMM7 được báo cáo vì các triệu chứng đòi hỏi phải có đột biến đồng hợp tử; các cá thể dị hợp tử chỉ mang biến thể và không biểu hiện bất kỳ đặc điểm lâm sàng bất thường nào. Cho đến nay, chỉ có 12 ca bệnh đột biến TOMM7 được ghi nhận, bao gồm 2 biến thể sai nghĩa gây bệnh (c.73T>C, p.Trp25Arg 5; c.86C>T, p.Pro29Leu 3) và một biến thể cắt nối hai alen (c.153-2A>C 7). Tầm vóc thấp và chậm phát triển là những triệu chứng phổ biến nhất liên quan đến tình trạng thiếu hụt TOMM7 do các biến thể giảm chức năng (hypomorphic variants) như c.73T>C, p.Trp25Arg 5 gây ra. Các biến thể mất chức năng hai alen làm gián đoạn hoạt động của phức hợp TOM sẽ gây ra tình trạng rối loạn chức năng ty thể nghiêm trọng và dẫn đến hội chứng Leigh khởi phát sớm 7. Trong số 12 ca bệnh được báo cáo, có 1 ca liên quan đến một người đàn ông gốc Hoa sống ở Malaysia, 9 ca là người Đài Loan, và 2 ca còn lại đến từ Nhật Bản và Ấn Độ. Báo cáo của chúng tôi liên quan đến một bé trai từ một gia đình người Hán ở Trung Quốc mang biến thể TOMM7 p.Pro29Leu; tất cả các bệnh nhân được biết đến cho đến nay đều có tổ tiên là người châu Á. Biến thể TOMM7 này cũng cho thấy tần suất alen cao hơn đáng kể ở các quần thể Đông Á (0,000652, 12/18.394 alen) so với tổng số toàn cầu (0,000048, 12/251.470 alen) trong Cơ sở dữ liệu tổng hợp bộ gen (gnomAD). Sự khác biệt tiềm ẩn về chủng tộc đối với tỷ lệ mắc GMPGS cần được nghiên cứu thêm.
Growth hormone is a peptide hormone secreted by the anterior lobe of the pituitary gland. It acts on nearly all body tissues, including bone, to promote growth in children. Recombinant hGH therapy treats short stature caused by several medical conditions, including growth hormone deficiency or insufficiency; birth small for gestational age; Prader-Willi syndrome 8; Turner syndrome 9; Noonan syndrome 10; and other rare genetic disorders. In our patient, various interventions were attempted, including a high-calorie diet, appetite enhancement, regulation of activity and rest patterns, and structured exercise. These approaches did not result in improvement. The child experienced severe growth arrest for 8 months before initiation of hGH therapy, accompanied by a period of weight loss. Growth hormone stimulation testing revealed partial growth hormone deficiency, and no contraindications to hGH treatment were identified 11. After initiation of growth hormone therapy at 31 months of age, the patient showed improved growth. His length increased, and his weight demonstrated a positive trend. Serum IGF-1 and IGF-BP3 levels also increased after hGH therapy began. No adverse reactions were observed during the treatment period. Notably, the previously elevated serum LDH and CK-MB levels – also observed by Young et al 5 in a patient with TOMM7 mutation – returned to normal in our patient. This improvement may be related to findings by Lakehal et al, who reported that growth hormone administration increases the oxidative capacity of red-type skeletal muscle 12, and by Nylander et al, who reported that growth hormone restores mitochondrial function and cellular membrane integrity 13.
Hormone tăng trưởng là một hormone peptide được bài tiết bởi thùy trước của tuyến yên. Nó tác động lên gần như tất cả các mô trong cơ thể, bao gồm cả xương, để thúc đẩy sự tăng trưởng ở trẻ em. Liệu pháp hGH tái tổ hợp điều trị chứng tầm vóc thấp do một số tình trạng bệnh lý gây ra, bao gồm thiếu hụt hoặc suy giảm hormone tăng trưởng; trẻ sinh ra nhỏ so với tuổi thai; hội chứng Prader-Willi 8; hội chứng Turner 9; hội chứng Noonan 10; và các rối loạn di truyền hiếm gặp khác. Ở bệnh nhân của chúng tôi, nhiều biện pháp can thiệp đã được thử nghiệm, bao gồm chế độ ăn nhiều calo, tăng cường cảm giác thèm ăn, điều chỉnh chế độ hoạt động và nghỉ ngơi, cũng như tập thể dục có cấu trúc. Những phương pháp này không mang lại sự cải thiện. Trẻ bị ngừng tăng trưởng nghiêm trọng trong 8 tháng trước khi bắt đầu liệu pháp hGH, kèm theo một giai đoạn sụt cân. Nghiệm pháp kích thích hormone tăng trưởng cho thấy tình trạng thiếu hụt hormone tăng trưởng một phần, và không có chống chỉ định nào đối với việc điều trị bằng hGH được xác định 11. Sau khi bắt đầu liệu pháp hormone tăng trưởng lúc 31 tháng tuổi, bệnh nhân đã cho thấy sự cải thiện về tăng trưởng. Chiều dài của trẻ tăng lên, và cân nặng biểu hiện một xu hướng tích cực. Nồng độ IGF-1 và IGF-BP3 trong huyết thanh cũng tăng lên sau khi bắt đầu liệu pháp hGH. Không có phản ứng bất lợi nào được quan sát thấy trong suốt thời gian điều trị. Đáng chú ý, nồng độ LDH và CK-MB huyết thanh tăng cao trước đó – cũng được Young và cộng sự 5 quan sát thấy ở một bệnh nhân đột biến TOMM7 – đã trở lại bình thường ở bệnh nhân của chúng tôi. Sự cải thiện này có thể liên quan đến các phát hiện của Lakehal và cộng sự, những người đã báo cáo rằng việc sử dụng hormone tăng trưởng làm tăng khả năng oxy hóa của cơ xương loại đỏ 12, và của Nylander và cộng sự, những người đã báo cáo rằng hormone tăng trưởng giúp phục hồi chức năng ty thể và tính toàn vẹn của màng tế bào 13.
Compared with other genetic syndromes, the growth response observed in our patient appears less pronounced than the typical first-year response to recombinant hGH treatment in Turner syndrome (0.54–0.58 SDS) or Prader-Willi syndrome (0.79–0.94 SDS) 14. Although the absolute length ΔSDS improvement in the present case was modest (−0.34 over 10 months), this result must be interpreted in the context of the patient’s severe pretreatment growth failure, which involved a decline of −4.11 SDS after birth and near-complete growth arrest from 24 to 31 months of age. The reversal from rapid deterioration to modest improvement represents a meaningful clinical achievement. The positive weight ΔSDS (+0.26) further underscores the overall anabolic benefit of the therapy. Importantly, the follow-up duration in this study was limited to 10 months. Further longitudinal studies are warranted to evaluate long-term efficacy, safety, and the potential impact on developmental trajectories and disease progression. In addition to growth hormone therapy, patients may benefit from a supplement regimen designed to support mitochondrial function, which warrants further investigation. Such a regimen could include coenzyme Q10, B vitamins, L-carnitine, and α-lipoic acid to enhance oxidative phosphorylation, antioxidant defense, and mitochondrial biogenesis 15.
So với các hội chứng di truyền khác, đáp ứng tăng trưởng được quan sát thấy ở bệnh nhân của chúng tôi có vẻ kém rõ rệt hơn so với đáp ứng điển hình trong năm đầu tiên điều trị bằng hGH tái tổ hợp trong hội chứng Turner (0,54–0,58 SDS) hoặc hội chứng Prader-Willi (0,79–0,94 SDS) 14. Mặc dù mức cải thiện ΔSDS chiều dài tuyệt đối trong trường hợp này là khiêm tốn (−0,34 trong 10 tháng), kết quả này phải được diễn giải trong bối cảnh bệnh nhân bị suy giảm tăng trưởng nghiêm trọng trước khi điều trị, bao gồm mức giảm −4,11 SDS sau khi sinh và tình trạng gần như ngừng tăng trưởng hoàn toàn từ 24 đến 31 tháng tuổi. Sự đảo ngược từ tình trạng suy giảm nhanh chóng sang cải thiện khiêm tốn đại diện cho một thành tựu lâm sàng có ý nghĩa. Chỉ số ΔSDS cân nặng tích cực (+0,26) càng nhấn mạnh thêm lợi ích đồng hóa tổng thể của liệu pháp này. Điều quan trọng là thời gian theo dõi trong nghiên cứu này bị giới hạn trong 10 tháng. Cần có thêm các nghiên cứu dọc tiếp theo để đánh giá hiệu quả, tính an toàn lâu dài, và tác động tiềm tàng đối với tiến trình phát triển cũng như sự tiến triển của bệnh. Ngoài liệu pháp hormone tăng trưởng, bệnh nhân có thể được hưởng lợi từ một phác đồ bổ sung được thiết kế để hỗ trợ chức năng ty thể, điều này cần được nghiên cứu thêm. Phác đồ như vậy có thể bao gồm coenzyme Q10, các vitamin nhóm B, L-carnitine, và α-lipoic acid để tăng cường quá trình phosphoryl hóa oxy hóa, phòng thủ chống oxy hóa và kích thích sinh sinh học ty thể 15.
Conclusions
Kết luận
We have described a 2-year-old boy with a homozygous mutation in the TOMM7 gene (NM_019059.5: Exon 1: c.86C>T [p.Pro29Leu]) who exhibited severe stunted physical growth. After administration of hGH, he resumed growth in both length and weight. These preliminary findings suggest that hGH treatment can improve growth velocity at an early age in patients with TOMM7 mutations. Nevertheless, interpretation of the present findings is limited by the short follow-up duration and the single-case nature of this report. Long-term follow-up and additional case reports are essential to confirm and strengthen these findings, which may ultimately guide clinical decision-making in this rare condition.
Chúng tôi đã mô tả một bé trai 2 tuổi mang đột biến đồng hợp tử trong gen TOMM7 (NM_019059.5: Exon 1: c.86C>T [p.Pro29Leu]) biểu hiện tình trạng chậm phát triển thể chất nghiêm trọng. Sau khi sử dụng hGH, trẻ đã tiếp tục tăng trưởng cả về chiều dài và cân nặng. Những phát hiện sơ bộ này gợi ý rằng điều trị bằng hGH có thể cải thiện tốc độ tăng trưởng ở độ tuổi nhỏ ở bệnh nhân có đột biến TOMM7. Tuy nhiên, việc diễn giải các phát hiện hiện tại bị hạn chế bởi thời gian theo dõi ngắn và tính chất báo cáo ca bệnh đơn lẻ. Theo dõi dài hạn và các báo cáo ca bệnh bổ sung là rất cần thiết để xác nhận và củng cố các phát hiện này, từ đó có thể hướng dẫn việc đưa ra quyết định lâm sàng trong tình trạng hiếm gặp này.
Reference
- Moehle EA, Shen K, Dillin A, Mitochondrial proteostasis in the context of cellular and organismal health and aging: J Biol Chem, 2019; 294(14); 5396-407
- Sekine S, Wang C, Sideris DP, Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1: Mol Cell, 2019; 73(5); 1028-1043.e5
- Garg A, Keng WT, Chen Z, Autosomal recessive progeroid syndrome due to homozygosity for a TOMM7 variant: J Clin Invest, 2022; 132(23); e156864
- Li CY, Chen LW, Tsai MC, Homozygous variant in translocase of outer mitochondrial membrane 7 leads to metabolic reprogramming and microcephalic osteodysplastic dwarfism with moyamoya disease: EBioMedicine, 2024; 110; 105476
- Young C, Batkovskyte D, Kitamura M, A hypomorphic variant in the translocase of the outer mitochondrial membrane complex subunit TOMM7 causes short stature and developmental delay: HGG Adv, 2023; 4(1); 100148
- Shi D, Qi M, Zhou L, Endothelial mitochondrial preprotein translocase Tomm7-Rac1 signaling axis dominates cerebrovascular network homeostasis: Arterioscler Thromb Vasc Biol, 2018; 38(11); 2665-77
- Yeole M, Majethia P, Siddiqui S, Bi-allelic splicing variant, c.153-2A > C in TOMM7 is associated with Leigh syndrome: Am J Med Genet A, 2025; 197(2); e63892
- Moix Gil E, Giménez-Palop O, Caixàs A, Treatment with growth hormone in the Prader-Willi syndrome: Endocrinol Diabetes Nutr, 2018; 65(4); 229-36
- Backeljauw P, Blair JC, Ferran JM, Early GH treatment is effective and well tolerated in children with Turner syndrome: NordiNet(R) IOS and Answer Program: J Clin Endocrinol Metab, 2023; 108(10); 2653-65
- Rodriguez F, Gaete X, Cassorla F, Etiology and treatment of growth delay in Noonan syndrome: Front Endocrinol (Lausanne), 2021; 12; 691240
- Kim JH, Chae HW, Chin SO, Diagnosis and treatment of growth hormone deficiency: A position statement from Korean Endocrine Society and Korean Society of Pediatric Endocrinology: Endocrinol Metab (Seoul), 2020; 35(2); 272-87
- Lakehal F, Hannah MJ, Crompton LA, Lomax MA, The effect of growth hormone on muscle enzyme activity and lactate dehydrogenase isoenzyme pattern: Biotechnology in Growth Regulation, 1989; 232, Oxford, Butterworth-Heinemann
- Nylander E, Zelleroth S, Nyberg F, The protective and restorative effects of growth hormone and insulin-like growth factor-1 on methadone-induced toxicity in vitro: Int J Mol Sci, 2018; 19(11); 3627
- Ross J, Fridman M, Kelepouris N, Factors associated with response to growth hormone in pediatric growth disorders: Results of a 5-year registry analysis: J Endocr Soc, 2023; 7(5); bvad026
- Tinker RJ, Lim AZ, Stefanetti RJ, McFarland R, Current and emerging clinical treatment in mitochondrial disease: Mol Diagn Ther, 2021; 25(2); 181-206
Có thể bạn quan tâm
-
Đặc điểm của các tổn thương giả cành cây ở giác mạc trong bệnh tăng tyrosine máu bẩm sinh tuýp 1 trên kính hiển vi đồng tiêu trong cơ thể và chụp cắt lớp quang học bán phần trước
In Vivo Confocal Microscopy and Anterior Segment Optical Coherence Tomography Features of Corneal Pseudodendritic Lesions in Hereditary Tyrosinemia Type 1
-
Hiệu quả của các thời điểm khác nhau khi bắt đầu sử dụng caffeine nhằm cải thiện chức năng hô hấp ở trẻ sinh non
Effects of different timings of caffeine initiation to improve respiratory function in preterm infants
-
Bạn biết tập thể dục tốt cho bạn – vậy tại sao việc thực hiện nó lại khó khăn đến vậy?
You know exercise is good for you – so why is it so hard to put it into practice?